
TAK-755 1 500 UI, poudre et solvant pour solution injectable, boîte de 1 flacon de poudre flacon de solvant ( set) de 5 ml
Dernière révision : 26/02/2024
Taux de TVA : 0%
Laboratoire exploitant : TAKEDA FRANCE
Source :

TAK-755 est indiqué dans le traitement enzymatique substitutif (TES) chez les patients agés de 12 ans et plus atteints de purpura thrombotique thrombocytopénique congénital (PTTc) dû à un déficit en ADAMTS13.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients pouvant mettre en jeu le pronostic vital.
Réactions d'hypersensibilité
Une hypersensibilité de type allergique, incluant des réactions anaphylactiques, peut survenir. Les patients doivent être informés des signes précoces de réactions d'hypersensibilité, incluant mais sans s'y limiter, tachycardie, oppression thoracique, respiration sifflante et/ou détresse respiratoire aiguë, hypotension, urticaire généralisée, prurit, rhino-conjonctivite, oedème de Quincke, léthargie, nausées, vomissements, paresthésie, agitation, qui peuvent évoluer vers un choc anaphylactique. En cas de signes et symptômes de réactions allergiques graves, il convient d'interrompre immédiatement l'administration de TAK-755 et de dispenser les soins appropriés au patient.
Immunogénicité
Comme pour toutes les protéines thérapeutiques, il existe un risque d'immunogénicité. Aucun anticorps neutralisant n'a été rapporté chez les patients traités par TAK-755 dans le cadre des essais cliniques sur le PTTc. Les patients peuvent développer des anticorps dirigés contre l'ADAMTS13r après un traitement par TAK-755, ce qui pourrait entraîner une diminution de la réponse à l'ADAMTS13r (voir la rubrique Propriétés pharmacodynamiques).
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du médicament administré doivent être clairement enregistrés.
Teneur en sodium
Ce médicament contient moins de 1 mmol de sodium (23 mg) par mL, c'est-à-dire essentiellement « sans sodium ».
Résumé du profil de sécurité
Effets indésirables observés lors des essais cliniques
Les effets indésirables les plus fréquents étaient les céphalées (28,6 %) et les nausées (14,3 %).
Les effets indésirables ont été rapportés dans une étude de phase 3 comparant TAK-755 à des plasmathérapies (plasma frais congelé [PFC], plasma traité par solvant/détergent [S/D], ou concentrés de FVW:FVIII, selon l'assignation de l'investigateur) et une étude de phase 3b (voir la rubrique Propriétés pharmacodynamiques). Aucun événement indésirable grave évalué comme étant lié à TAK-755 n'a été observé dans ces essais cliniques.
Au cours des périodes 1, 2 et 3 de l'étude de phase 3, aucun patient recevant TAK-755 n'a présenté d'événements indésirables entraînant l'arrêt ou l'interruption du traitement, tandis qu'un patient sur 44 (2,3%) recevant des thérapies à base de plasma a présenté un événement indésirable entraînant l'arrêt du traitement et que 7 patients sur 44 (15,9%) ont présenté un total de 8 événements indésirables entraînant une interruption du traitement durant les traitements à base de plasma.
Les effets indésirables (EI) sont énumérés dans le tableau 1.
La
convention suivante est utilisée pour définir les catégories de
fréquences et est basée sur les lignes directrice du Conseil des
Organisations Internationales des Sciences Médicales (CIOMS) : : très
fréquent (≥1/10), fréquent (≥1/100 à <1/10), peu fréquent (≥1/1 000
à <1/100), rare (≥1/10 000 à <1/1 000), très rare (<1/10 000).
Au sein de chaque classe de systèmes d'organes (SOC), les effets
indésirables sont présentés par ordre décroissant de fréquence.
Tableau 1 : Effets indésirables rapportés chez >2 % des patients traités par TAK-755
| Classe de systèmes d'organes MedDRA(SOC) | Effet indésirable par Terme préférentiel (PT) | Catégorie de fréquence par patient |
| Affections du système nerveux | Céphalée | Très fréquent |
| Somnolence | Fréquent | |
| Affections vasculaires | Hypertension | Fréquent |
| Affections gastro-intestinales | Nausée | Très fréquent |
| Affections de la peau et du tissus sous-cutané | Prurit | Fréquent |
| Troubles généraux et anomalies au site d'administration | Sensation de chaud | Fréquent |
| Affections respiratoires, thoraciques et médiastinales | Infection des voies aériennes supérieures | Très fréquent |
Population pédiatrique
Il existe peu d'informations issues d'études contrôlées sur TAK-755 chez des patients pédiatriques.
La fréquence, le type et la gravité des effets indésirables chez les enfants devraient être les mêmes que chez les adultes.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté selon les modalités définies dans le Protocole d'utilisation thérapeutique et de recueil de données (voir PUT RD).
SURVEILLANCE pendant le traitement :
- Apparition des anticorps dirigés contre l'ADAMTS13r, ce qui pourrait entraîner une diminution de la réponse au traitement.
- La survenue d'hypersensibilité de type allergique et INFORMER les patients des signes et symptômes de ces réactions.
Grossesse
Il n'existe pas de données provenant d'essais cliniques contrôlés sur l'utilisation de l'ADAMTS13r chez les femmes enceintes.
Au cours du développement de TAK-755, trois patientes atteintes de PTTc ont été exposées à TAK- 755 pendant la grossesse.
Une patiente de l'étude 3002 s'est avérée enceinte environ une semaine après sa dernière dose de TAK-755. Elle a été retirée de l'étude pour se conformer aux exigences du protocole.
Environ deux mois après l'arrêt de l'étude, la patiente a fait une fausse couche au premier trimestre. L'investigateur a évalué que cet événement n'était pas lié à TAK-755.
Deux patientes atteintes de PTTc ont été traitées par TAK-755 dans le cadre d'un programme d'utilisation compassionnel pendant la grossesse. :
-
La première patiente, au cours du troisième trimestre de sa deuxième grossesse, a présenté un accident vasculaire cérébral et une thrombocytopénie réfractaire à la plasmaphérèse quotidienne. À 33 semaines de grossesse, le traitement par TAK-755 a été instauré une fois par semaine. Les niveaux d'activité d'ADAMTS13 se sont normalisés, la thrombocytopénie a été résolue et un nouveau-né en bonne santé est né à 37 semaines sans qu'aucun problème de sécurité n'ait été signalé par le médecin suivant la patiente comme lié à TAK-755.
-
La deuxième patiente a eu une exacerbation de son PTTc au cours de son deuxième trimestre de grossesse malgré un échange plasmatique quotidien antérieur. Sa grossesse a été considérée comme étant à risque, avec une réponse inadéquate aux thérapies à base de plasma. TAK-755 a été instauré une fois par semaine et a induit une rémission clinique. Le nouveau-né est né par césarienne à 29 semaines et le médecin n'a signalé aucun effet indésirable lié à TAK-755.
Les études chez l'animal n'indiquent pas de toxicité sur la reproduction (voir la rubrique Données de sécurité préclinique).
L'utilisation de TAK-755 pendant la grossesse, si cela est nécessaire, ne peut être envisagée qu'après une analyse individuelle approfondie des risques et des bénéfices par le médecin suivant la patiente avant et pendant le traitement. L'utilisation de TAK-755 pendant la grossesse est une décision qui relève exclusivement de l'appréciation médicale du médecin.
Allaitement
On ne sait pas si l'ADAMTS13r est présent dans le lait maternel, affecte la lactation ou a des effets sur le nourrisson allaité. Les avantages de l'allaitement sur le développement et la santé doivent être pris en compte, de même que la nécessité pour la mère d'utiliser TAK-755 et tout effet indésirable potentiel pour l'enfant allaité.
Fertilité
Les effets de l'ADAMTS13r sur la fertilité chez l'homme n'ont pas été établis. Une étude sur la fertilité des femelles et le développement embryo-foetal chez le rat n'a montré aucun effet maternel indésirable ni aucun effet lié au traitement sur la fertilité à des doses allant jusqu'à 400 U/kg. Aucun effet sur les paramètres séminaux ou sur les organes reproducteurs n'a été observé chez les rats à des doses allant jusqu'à 400 U/kg (voir la rubrique Données de sécurité préclinique).
Aucune étude d'interaction n'a été réalisée avec TAK-755.
Le traitement par TAK-755 doit être instauré sous la supervision d'un médecin expérimenté dans la prise en charge des patients atteints de troubles hématologiques.
Posologie
Traitement enzymatique substitutif prophylactique
· 40 UI/kg de poids corporel de TAK-755 doivent être administrés une fois toutes les deux semaines.
· La fréquence d'administration de la prophylaxie peut être ajustée à 40 UI/kg de poids corporel une fois par semaine en fonction du schéma posologique prophylactique antérieur ou de la réponse clinique (voir les rubriques Propriétés pharmacodynamiques, Propriétés pharmacocinétiques).
Traitement enzymatique substitutif à la demande
En cas d'épisode aigu de PTT, la posologie recommandée de TAK-755 pour traiter les épisodes aigus de PTT est la suivante :
· 40 UI/kg de poids corporel de TAK-755 doivent être administrées le jour 1.
· 20 UI/kg de poids corporel de TAK-755 doivent être administrées le jour 2.
· 15 UI/kg de poids corporel de TAK-755 doivent être administrées à partir du jour 3, une fois par jour, jusqu'à deux jours après la résolution de l'épisode aigu (voir les rubriques Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Populations particulières
Personnes âgées
Les données sur l'utilisation de TAK-755 chez les patients de plus de 65 ans sont limitées. Sur la base des résultats d'analyse pharmacocinétique, aucun ajustement posologique n'est nécessaire chez les patients âgés (voir la rubrique Propriétés pharmacocinétiques).
Insuffisance rénale
Aucun ajustement posologique de TAK-755 n'est nécessaire chez les patients présentant une insuffisance rénale légère ou modérée (voir la rubrique Propriétés pharmacocinétiques). L'administration de TAK-755 chez les patients atteints d'insuffisance rénale terminale (IRT) nécessitant une dialyse chronique n'a pas été étudiée.
Insuffisance hépatique
Aucun ajustement posologique de TAK-755 n'est nécessaire chez les patients présentant une insuffisance hépatique légère (classe A de Child-Pugh) ou modérée (classe B de Child-Pugh) (voir la rubrique Propriétés pharmacocinétiques). L'administration de TAK-755 chez les patients présentant un dysfonctionnement hépatique n'a pas été étudiée.
Population pédiatrique
Le schéma posologique recommandé en fonction du poids corporel chez les patients pédiatriques est le même que chez les adultes.
Il existe peu d'informations issues d'études contrôlées sur TAK-755 chez des patients pédiatriques. Les expositions à l'activité d'ADAMTS13 devraient être similaires chez les adultes, les adolescents et les patients pédiatriques (voir les rubriques Effets indésirables, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Mode d'administration
A utiliser par voie intraveineuse après reconstitution uniquement.
TAK-755 500 UI et TAK-755 1500 UI, poudre et solvant pour solution injectable, est administré à raison de 2 à 4 mL par minute.
Administration à domicile
L'administration à domicile sous la supervision d'un professionnel de santé peut être envisagée pour les patients qui tolèrent bien leurs perfusions. La décision de faire passer un patient à la perfusion à domicile doit être prise après évaluation et recommandation du médecin suivant le patient. La dose et le débit de perfusion doivent rester constants à domicile et ne pas être modifiés sans avis du médecin. Si le patient présente des signes précoces d'hypersensibilité pendant la perfusion à domicile, le processus de perfusion doit être arrêté immédiatement et un traitement approprié doit être instauré (voir la rubrique Mises en garde spéciales et précautions d'emploi). Il se peut que les perfusions suivantes doivent être effectuées dans un établissement clinique. Le traitement doit être étroitement surveillé par le médecin suivant le patient.
Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique Précautions particulières d'élimination et de manipulation.
Durée de conservation :
Flacon non ouvert 3 ans
Durée de conservation après reconstitution
La stabilité physico-chimique en cours d'utilisation a été démontrée pendant 3 heures à 25 °C.
D'un point de vue microbiologique, sauf si la méthode de reconstitution
prévient tout risque de contamination microbienne, le produit doit être
utilisé immédiatement. En cas d'utilisation non immédiate, les durées
et conditions de conservation en cours d'utilisation relèvent de la
responsabilité de l'utilisateur.
À conserver au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler.
A conserver dans l'emballage d'origine, à l'abri de la lumière.
TAK-755
peut être conservé à température ambiante jusqu'à 30°C pendant une
période allant jusqu'à 6 mois sous forme lyophilisée, mais sans
dépasser la date de péremption.
Ne pas remettre TAK-755 dans au réfrigérateur après l'avoir conservé à température ambiante.
Noter sur la boîte la date à laquelle TAK-755 est retiré du réfrigérateur.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Il n'existe pas de données sur le surdosage de TAK-755 chez les patients. En cas de surdosage, sur la base de l'action pharmacologique de l'ADAMTS13r, il existe un risque potentiel accru de saignement (voir la rubrique Propriétés pharmacodynamiques).
Classe pharmacothérapeutique : non encore attribué
Mécanisme d'action
L'ADAMTS13 est une métalloprotéase plasmatique à zinc qui régule l'activité du facteur de von Willebrand (FVW) en clivant les multimères FVW de haut et de très haut poids moléculaire en unités plus petites et en réduisant ainsi les propriétés de liaison plaquettaire du FVW et sa capacité à former des microthrombi. TAK-755 est une forme recombinante de l'ADAMTS13 endogène avec des propriétés pharmacocinétiques similaires. L'utilisation de TAK-755 chez les patients atteints de PTTc fournit une supplémentation ciblée en ADAMTS13 et une reconstitution de l'activité plasmatique d'ADAMTS13, ce qui devrait réduire ou éliminer la formation spontanée de microthrombi de plaquettes-FVW qui conduit à la consommation de plaquettes et à la thrombocytopénie, étant un marqueur de l'activité de la maladie chez les patients atteints de PTTc.
Effets pharmacodynamiques
L'activité du cofacteur de la ristocétine (FVW:RCo) a été utilisée pour évaluer l'activité du FVW. Après des doses intraveineuses (IV) de TAK-755 à la dose recommandée, l'antigène FVW et le FVW:RCo ont diminué de façon transitoire pendant 1 à 2 jours, avec un changement de 15 % à 25 % par rapport à la valeur initiale.
Efficacité et sécurité clinique
L'efficacité clinique et la sécurité ont été évaluées dans le cadre de deux essais cliniques en cours (étude 281102 et étude 3002).
Étude 281102 (étude pivot)
TAK-755 a fait l'objet d'une étude internationale de phase 3, prospective, randomisée, contrôlée, ouverte, multicentrique, croisée sur deux périodes (cross-over), suivie d'une période de continuation mono-bras (étude 281102) évaluant l'efficacité et la sécurité de TES prophylactique et à la demande ou aiguë avec TAK-755 par rapport aux plasmathérapies chez les patients atteints de PTTc.
Traitement prophylactique enzymatique substitutif chez les patients atteints de PTTc
L'efficacité de TAK-755 dans le traitement prophylactique des patients atteints de PTTc a été évaluée chez 46 patients de la cohorte prophylactique qui ont été randomisés pour recevoir 6 mois de traitement prophylactique avec soit 40 UI/kg (±4 UI/kg) de TAK-755, soit des plasmathérapies (période 1), puis sont passés à l'autre traitement pendant 6 mois (période 2). Trente-cinq patients sont entrés ensuite dans un bras unique pour une période de traitement de 6 mois avec TAK-755 (Période 3).
L'âge moyen (écart-type) était de 30,5 (16,0) ans (intervalle : 3 à 58 ans), le poids moyen (écart-type) était de 65,9 kg (21,8) (intervalle : 18,5 à 102,4 kg), et la majorité des patients étaient blancs (65,2 %), non hispaniques ou latinos (80,4 %) et étaient des femmes (58,7 %), dont 74,1 % étaient en âge de procréer.
Huit patients (17,4 %) ont présenté au moins un épisode aigu de PTT au cours des 12 mois précédant le recrutement. Avant de rejoindre l'étude, la majorité (69,6 %) des patients ont reçu un traitement par PFC, 21,7 % ont reçu du plasma S/D et 6,5 % ont reçu du concentré FVW/FVIII.
L'efficacité du traitement prophylactique avec TAK-755 chez les patients atteints de PTTc a été démontrée sur la base de l'incidence des épisodes aigus de PTT, des épisodes subaigus de PTT et des manifestations de PTT ; ainsi que toute dose supplémentaire administrée en raison d'évènements de PTT subaigus.
Aucun patient recevant TAK-755 n'a eu d'épisode aigu de PTT tout au long de l'étude, y compris pendant la période 3 (avec une durée médiane d'exposition à TAK-755 de 14 mois pour les patients âgés de 12 à <18 ans et les patients âgés de ≥18 ans ; et 4 et 1 mois chez les patients âgés de 6 à <12 ans et <6 ans, respectivement). Un épisode aigu de PTT est survenu chez un patient recevant du PFC en prophylaxie en période 1(voir tableau 2).
Aucun épisode de PTT subaigu n'a été rapporté chez les patients recevant TAK-755 au cours des périodes 1 et 2. Au cours de la période 3, deux patients recevant une prophylaxie par TAK-755 ont présenté deux épisodes subaigus, dont l'un a nécessité quatre doses supplémentaires. Quatre patients recevant une plasmathérapie ont présenté cinq épisodes subaigus de PTT au cours des périodes 1 et 2. Au total, six doses supplémentaires ont été administrées à deux de ces patients.
Les patients recevant TAK-755 ont présenté un taux annualisé de manifestations de PTT inférieur à celui des plasmathérapies, à l'exception des manifestations de PTT liées à un dysfonctionnement rénal. L'incidence annualisée des manifestations de thrombocytopénie a diminué de 60 % (valeur nominale de p = 0,002) sous TAK-755, en comparaison aux plasmathérapies (voir tableau 2).
Tableau 1: Résultats de l'efficacité de le cohorte prophylactique chez les patients atteints de PTTc âgés de ≥ 12 ans (périodes 1 et 2)
| | [TAK-755] N=37 | Plasmathérapie N=38 |
| Épisodes aigus de PTTa | | |
| Nombre de patients (nombre d'épisodes) | 0(0) | 1(1) |
| Taux moyen d'épisodes annualisé non basé sur un modèle (ET)b | 0 (0.000) | 0.05 (0.280) |
| Épisodes subaigus de PTTc | | |
| Nombre de patients (nombre d'épisodes) | 0(0) | 4(5) |
| Taux moyen d'épisodes annualisé non basé sur un modèle (ET)b | 0 (0.000) | 0.25 (0.778) |
| Manifestations de PTT | | |
| Thrombocytopénied | | |
| Nombre de patients (nombre d'événements) | 9(30) | 19(75) |
| Taux d'événements annualisé basé sur un modèlee MMC (ES) | 0,74 (0,257)* | 1.73 (0.533) |
| Anémie hémolytique microangiopathiquef | | |
| Nombre de patients (nombre d'événements) | 5(7) | 11(20) |
| Taux d'événements annualisé basé sur un modèlee MMC (ES) | 0.26 (0.130) | 0.74 (0.244) |
| Symptômes neurologiquesg | 4(18) | 7(29) |
| Nombre de patients (nombre d'événements) |
| Taux d'événements annualisé basé sur un modèlee MMC (ES) | 0.18 (0.093) | 0.29 (0.142) |
| Dysfonctionnement rénalh | | |
| Nombre de patients (nombre d'événements) | 3(8) | 2(5) |
| Taux d'événements annualisé basé sur un modèlee MMC (ES) | 0.10 (0.078) | 0.08 (0.059) |
| Douleurs abdominales | | |
| Nombre de patients (nombre d'événements) | 2(4) | 5(7) |
| Taux d'événements annualisé basé sur un modèlee MMC (ES) | 0.11 (0.068) | 0.17 (0.094) |
| Autres manifestations du PTTi | | |
| Nombre de patients (nombre d'événements) | 6(9) | 14(24) |
| Taux d'événements annualisé basé sur un modèlee MMC (ES) | 0,32 (0,127)* | 0.81 (0.244) |
| Manifestations composites du PTT (à l'exclusion des autres manifestations du PTT) | | |
| Nombre de patients (nombre d'événements) | 18(66) | 24(117) |
| Taux d'événements annualisé basé sur un modèlee MMC (ES) | 2,28 (0,596)* | 3.68 (0.910) |
MMC = méthode des moindres carrés ; ET = écart type ; ES = erreur-standard ; PTT = purpura thrombotique thrombocytopénique.
a Les épisodes aigus de PTT ont été définis par une baisse de la numération plaquettaire (≥50 % de la valeur initiale ou une numération plaquettaire <100 000/µL) et une élévation de la lactate déshydrogénase (LDH) (>2× la valeur initiale ou >2× la limite supérieure normale (LSN)).
b Taux d'épisodes annualisé non modélisé pour un sujet = nombre d'épisodes/durée de la période d'observation (années).
c Les épisodes subaigus ont été définis par un événement de thrombocytopénie ou d'anémie hémolytique microangiopathique ainsi que par des signes et symptômes spécifiques aux organes, y compris, mais sans s'y limiter, des événements de dysfonctionnement rénal, des symptômes neurologiques, de la fièvre, de la fatigue/léthargie et/ou des douleurs abdominales.
d Les événements de thrombocytopénie ont été définis comme une baisse de la numération plaquettaire ≥25 % de la valeur initiale ou une numération plaquettaire <150 000/µL.
e D'après un modèle binominal négatif à effets mixtes avec traitement (TAK-755 vs plasmathérapies), la période de traitement (1 et 2) et séquence (plasmathérapies-TAK-755 vs TAK-755-plasmathérapies) comme effet fixe et patient comme effet aléatoire. En raison de la rareté des événements, les résultats pour les événements de dysfonction rénale n'incluaient pas la séquence de traitement comme covariable.
f Les anémies hémolytiques microangiopathiques ont été définies comme une élévation de la LDH >1,5× la valeur initiale ou >1,5 x LSN.
g Les symptômes neurologiques comprenaient les symptômes de troubles du système nerveux liés au PTT (p. ex. céphalées, confusion, troubles de la mémoire, irritabilité, paresthésie, dysarthrie, dysphonie, troubles visuels, symptômes moteurs focaux ou généraux dont les convulsions).
h Les événements de dysfonction rénale ont été définis comme une augmentation de la créatinine sérique >1,5× la valeur initiale.
i Les autres manifestations de PTT comprenaient tous les effets indésirables signalés par les investigateurs qui étaient considérés comme liés ou possiblement liés au PTTc et qui n'étaient pas considérés comme des symptômes neurologiques, des douleurs abdominales, une thrombocytopénie, une augmentation de la LDH ou une augmentation de la créatinine.
* Valeur nominale de p <0,05 pour la différence de traitement dans les taux d'événements annualisés basés sur le modèle.
Les résultats globaux d'efficacité de TAK-755 ont été cohérents tout au long de l'étude, y compris à la période 3 et dans tous les groupes d'âge.
La satisfaction à l'égard du traitement et la qualité de vie liée à la santé (QVLS) ont également été évaluées au cours de cette étude. Les résultats rapportés par les patients (PRO), basés sur des questionnaires pertinents, ont été rapportés à l'inclusion et à la fin de chaque période pour la cohorte prophylactique. Dans l'ensemble, les symptômes, les impacts et les scores QVLS liés à la maladie déclarés par les patients sont restés cohérents entre les différents moments de l'étude (inclusion, fin de la période 1 et fin de la période 2) pour les patients recevant TAK-755 et des plasmathérapies. Les patients recevant TAK-755 ont systématiquement rapporté des scores plus élevés dans les domaines de la satisfaction du traitement, évalués à l'aide du questionnaire de satisfaction du traitement pour les médicaments (TSQM-9), par rapport aux patients traités avec des plasmathérapies et par rapport aux scores à l'inclusion indiquant une efficacité, une commodité et une satisfaction perçues plus élevées à l'égard de TAK-755.
Traitement enzymatique substitutif à la demande ou en phase aiguë
L'efficacité de la thérapie enzymatique substitutive à la demande (OD) ou aiguë a été évaluée en fonction de la proportion d'épisodes aigus de PTT répondant à TAK-755 dans les cohortes prophylactiques et OD pendant toute la durée de l'étude.
Un épisode aigu de PTT répondant à TAK-755 a été défini comme un épisode résolu lorsque la numération plaquettaire était ≥150 000/µL ou que la numération plaquettaire se situait à moins de 25 % de la valeur initiale, selon la première éventualité, et que la LDH ≤1,5 x la valeur initiale ou ≤1,5 x la LSN, sans nécessiter l'utilisation d'un autre actif contenant de l'ADAMTS13.
Cinq patients adultes (âgés de ≥18 ans) ont été inclus dans la cohorte OD et ont présenté un total de six épisodes aigus de PTT. Parmi ces cinq patients, deux patients ont été randomisés pour recevoir un traitement à la demande avec TAK-755 et trois patients ont été randomisés pour recevoir des plasmathérapies. Les 6 épisodes aigus de PTT ont disparu après un traitement par TAK-755 ou par des plasmathérapies.
La plupart des patients (60%) étaient des hommes, blancs (60%) avec un âge médian (min, max) de 20 (20, 36) ans, un poids moyen (ET) de 63,1 (10,0) kg et un poids médian (min, max) de 65 (46,8, 74,0) kg. Aucun des patients n'était hispanique ou latino.
Étude 3002 (étude de continuation)
Les patients qui ont terminé l'étude de phase 3 (étude 281102) étaient éligibles pour participer à une étude de continuation de phase 3b, prospective, ouverte, multicentrique, à traitement unique (étude 3002) évaluant la sécurité et l'efficacité de TAK-755 dans le traitement prophylactique et à la demande des patients atteints de PTTc.
Trente-six patients ont reçu un traitement prophylactique TAK-755 (dont 33 patients venant de l'étude 281102). Aucun patient n'a été inclus dans la cohorte à la demande. Les taux d'incidence des épisodes aigus et subaigus de PTT et des manifestations du PTT restent cohérents avec les résultats observés dans l'étude 281102 avec des durées moyennes et maximales de traitement de 0,58 an et 1,4 an, respectivement.
Immunogénicité
Tous les sujets ont fait l'objet d'un suivi de la présence d'anticorps neutralisants (inhibiteurs) dirigés contre l'ADAMTS13r dans le cadre d'essais cliniques sur le PTTc. Aucun patient n'a développé d'anticorps neutralisants dirigés contre l'ADAMTS13r.
Six des 54 patients traités par TAK-755 avec un PTTc confirmé ont été testés positifs pour les anticorps de liaison à faible titre contre l'ADAMTS13r sans impact clinique observable sur l'innocuité ou l'efficacité de TAK-755, et sans augmentation des titres d'anticorps au fil du temps. En raison de la faible occurrence d'ADA, l'effet des anticorps de liaison sur la pharmacocinétique, la pharmacodynamie, la sécurité et/ ou l'efficacité des produits ADAMTS13r ne peut pas être évalué (voir la rubrique Mises en garde spéciales et précautions d'emploi).
L'Agence Européenne des médicaments a reporté l'obligation de soumettre les résultats des études menées avec TAK-755 dans un ou plusieurs sous-ensembles de la population pédiatrique dans le traitement du purpura thrombotique thrombocytopénique, conformément à la décision relative au plan d'investigation pédiatrique (PIP). Cependant, les résultats des patients pédiatriques ont été inclus dans la demande d'autorisation de mise sur le marché pour l'indication accordée (voir les rubriques Posologie et mode d'administration, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques pour des informations sur l'utilisation pédiatrique).
Le profil pharmacocinétique de TAK-755 a été déterminé sur la base de l'activité d'ADAMTS13 analysée lors des essais cliniques.
Après l'administration intraveineuse d'une dose unique de TAK-755 à 5 UI/kg, 20 UI/kg et 40 UI/kg à des adultes et des adolescents, des augmentations liées à la dose de l'activité ADAMTS13 individuelle ont été observées et ont atteint un maximum environ 1 heure après la perfusion ou plus tôt. À la dose clinique de 40 UI/kg, la demi-vie moyenne (écart-type (ET)) et le temps de résidence moyen (TRM) chez les adultes et les adolescents étaient respectivement de 47,8 (13,7) heures et 63,8 (16,0) heures.
Les paramètres pharmacocinétiques de l'activité d'ADAMTS13, après l'administration IV de TAK-755 à 40 UI/kg chez les adultes et les adolescents sont décrits dans le Tableau 3.
Tableau 2 : Paramètres pharmacocinétiques de l'activité ADAMTS13 après l'administration IV de TAK-755 chez les patients atteints de PTTc ≥ 12 ans
| Paramètre (unité) | (ET) Min; Max (N=23) |
| Cmax (UI/mL) | 1.15 (0.25)0.78; 1.56 |
| IR[(UI/mL)/(UI/kg)] | 0.03 (0.01)0.02; 0.04 |
| AUCall (UI*h/mL) | 52.8 (12.9)34.0; 84.0 |
| TRM0-infa (h) | 63.8 (16.0)44.8; 113 |
| C moyenne (0-168 h)(UI/mL) | 0.30 (0.07)0.20; 0.46 |
| Durée d'activité d'ADAMTS13 supérieure à 10%(jours) | 5.8 (1.2)4.5; 8.9 |
AUC = aire sous la courbe activité-temps ; Cmoyenne (0-168 h) = activité moyenne ADAMTS13 de 0 à 168 heures d'intervalle de dosage ; Cmax = activité maximale ADAMTS13 ; IR = récupération incrémentielle ; TRM = temps de résidence moyen.
Remarque : 1 UI/ml ADAMTS13 d'activité correspond à une activité normale moyenne de 100 %.
a N=22
Remarque : 1 UI/ml ADAMTS13 d'activité correspond à une activité normale moyenne de 100 %.
a N=22
L'administration IV de TAK-755 à 40 UI/kg a entraîné des expositions à une activité ADAMTS13 environ plus de 5 fois plus élevée (Cmax, AUC et durée supérieure à 10 % d'activité d'ADAMTS13) et une variabilité plus faible par rapport aux thérapies plasmatiques.
Sur la base de l'analyse pharmacocinétique de population exploitant les données disponibles (N = 65) de patients adultes, adolescents et pédiatriques, la Cmax moyenne (ET) à l'état d'équilibre, l'AUCall, la Cmoyenne et la durée d'activité d'ADAMTS13 supérieure à 10% après l'administration IV de TAK-755 toutes les deux semaines chez les patients atteints de PTTc de moins de 12 ans étaient de 1,02 (0,19) UI/mL, 54,0 (12,2) UI*h/mL, 0,16 (0,04) UI/ml et 6,8 (2,1) jours, respectivement.
L'antigène de l'ADAMTS13 et les caractéristiques pharmacocinétiques (TRM, Vss (volume de distribution à l'état d'équilibre) et CL) étaient similaires dans tous les groupes d'âge chez les patients atteints de PTTc. La posologie de TAK-755 basée sur le poids corporel fournit des paramètres pharmacocinétiques d'activité ADAMTS13 similaires (Cmax et Cmoyenne) dans les différents groupes d'âge, y compris les patients pédiatriques âgés de <12 ans.
Populations spéciales
Âge, sexe, race et autres facteurs intrinsèques
Outre le schéma posologique basé sur le poids corporel, aucun ajustement posologique n'est nécessaire car aucun facteur intrinsèque tel que l'âge, le sexe, la race, le débit de filtration glomérulaire estimé (DFGe) à l'inclusion et la bilirubine à l'initiation n'a été identifié comme covariables ayant une incidence sur la pharmacocinétique de l'ADAMTS13r.
Population pédiatrique
L'utilisation de TAK-755 chez les patients de moins de 12 ans est également supportée par les éléments suivants :
-
Données issues d'études contrôlées portant sur TAK-755 chez des patients âgés de 12 ans et plus (voir la rubrique Propriétés pharmacodynamiques),
-
Analyse pharmacocinétique (PK) de population utilisant les données disponibles chez les adultes, les adolescents et les patients pédiatriques de moins de 12 ans (N = 65) démontrant qu'en dehors de la posologie reliée au poids corporel, aucun ajustement posologique supplémentaire n'est nécessaire pour tenir compte de l'âge (voir la rubrique Propriétés pharmacocinétiques),
-
L'évolution de la maladie devrait être similaire entre les adultes, les adolescents et les patients pédiatriques de moins de 12 ans, ce qui permet l'extrapolation des données des adultes aux patients pédiatriques.
rADAMTS13 peut avoir une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Des vertiges/étourdissements et une somnolence peuvent survenir suite à l'administration d'apadamtase alfa (voir rubrique Effets indésirables).
Cancérogenèse, Mutagenèse
Aucune étude n'a été menée avec la substance active de TAK-755 pour évaluer son potentiel mutagène ou cancérigène.
Toxicologie et pharmacologie animales
TAK-755 clive le FVW de souris, rat, cochon d'Inde, singe et cochon nain. L'administration prophylactique ou thérapeutique de TAK-755 s'est avérée efficace dans le PTT induit par FVWr chez des souris knock-out (KO) ADAMTS13 d'une manière dose-dépendante (1-200 U/kg) qui dépendait également de l'intervalle entre le traitement par TAK-755 et l'induction par FVWr. Dans un modèle de saignement de l'extrémité de la queue du rat avec une pharmacologie exagérée à des niveaux supra- physiologiques de TAK-755, le risque d'augmentation des événements hémorragiques était faible.
L'administration de TAK-755 à des rats tous les trois jours par injection intraveineuse à 80, 400 ou 800 U/kg pendant 28 jours (10 administrations) a été bien tolérée et n'a provoqué aucun effet indésirable lié au produit. De plus, l'administration de TAK-755 à des rats par injection intraveineuse une fois par jour à 800 ou 1820 UI/kg pendant 30 jours a été bien tolérée sans aucun signe de toxicité. L'administration de TAK-755 à des rats tous les trois jours par injection intraveineuse à 80, 200 ou 400 U/kg pendant 26 semaines a été bien tolérée, sans signe de toxicité. TAK-755 n'a eu aucun effet sur les paramètres séminaux ni sur les organes reproducteurs mâles et femelles des rats étudiés.
Dans une étude de toxicité à doses répétées de 4 semaines chez le singe, l'administration de TAK-755 à 80, 200 ou 400 U/kg n'a pas entrainé de signes cliniques indésirables ou de résultats pouvant être directement attribués au produit. Dans une autre étude, l'administration de TAK-755 à 800 U/kg de poids corporel chez des singes cynomolgus une fois par semaine pendant 4 semaines n'a pas entrainé d'effets indésirables. Les titres d'anticorps neutralisants ont été nettement augmentés chez certains singes, ce qui a entraîné une anémie hémolytique et une thrombocytopénie. Ces changements ont été considérés comme étant probablement causés par le développement d'anticorps neutralisants à réaction croisée contre l'ADAMTS13 endogène du singe. La formation d'anticorps chez les animaux n'est pas considérée comme prédictive de la formation d'anticorps chez l'homme.
Dans une étude de faisabilité de 2 semaines chez le lapin, l'administration de 80, 400 ou 800 U/kg IV de TAK-755 a entraîné une diminution de l'hématocrite, de la concentration d'hémoglobine et de la numération plaquettaire, ainsi que des hémorragies à la dose de 800 U/kg. Une dégénérescence/nécrose des myocytes du coeur et une inflammation aiguë/subaiguë ont été observées dans le myocarde avec une distribution locale/diffuse à 400 U/kg ou 800 U/kg. Ces résultats sont évocateurs d'un PTT, qui serait attribué aux anticorps anti-médicaments (ADA)/ neutralisants qui se forment contre TAK-755. Le lapin n'a pas été considéré comme une espèce appropriée pour les essais de toxicité en raison de l'incidence élevée d'anticorps anti-médicaments (ADA)/anticorps neutralisants et du manque d'activité pharmacologique.
Toxicologie de la reproduction
Dans une étude sur la fertilité des femelles et le développement embryo-foetal chez le rat, TAK-755 a été administré tous les trois jours pendant 2 semaines avant l'accouplement, pendant l'accouplement, et jusqu'au 16e jour environ de gestation à 80, 200 ou 400 U/kg chez les rats femelles et n'a pas été associé à des signes maternels indésirables ou à des effets liés au traitement sur la fertilité, sur la gestation, ou sur le développement foetal. Dans l'étude de toxicité chronique, aucune préoccupation pour la fertilité masculine ou féminine n'a été identifiée (voir Toxicologie animale et pharmacologie). TAK-755 a été administré à des doses de 80, 200 ou 400 U/kg par injection intraveineuse (IV) en bolus tous les trois jours, du jour 6 de la gestation au sevrage vers le 21e jour de lactation dans une étude prénatale et postnatale chez le rat. Il n'y avait aucun signe ou effet clinique indésirable sur la prise de poids corporel, la consommation alimentaire ou les paramètres hématologiques chez la mère ou la progéniture 1ère génération (F1) ou 2ème génération (F2). Aucune étude de toxicité juvénile n'a été menée avec TAK-755, car aucun effet indésirable n'a été observé chez les rats adultes (études de toxicité et de développement à doses répétées et de toxicité pour la reproduction).
En raison de son haut poids moléculaire, ADAMTS13r devrait avoir un passage transplacentaire limité. Dans les études sur les rats, il n'y a pas eu de passage transplacentaire biologiquement significatif, ni de toxicité sur la reproduction ou le développement à des doses allant jusqu'à 400 U/kg, ni d'effet sur la fertilité à des doses allant jusqu'à 400 U/kg.
TAK-755 doit être administré par voie intraveineuse après reconstitution de la poudre avec l'eau pour préparations injectables fournie.
Instructions générales
- Calculer la dose et le volume d'administration en fonction du poids corporel du patient
- Utiliser une technique aseptique tout au long de la procédure.
- Vérifier la date d'expiration du produit avant utilisation.
- Ne pas utiliser TAK-755 si la date d'expiration est dépassée.
- Si le patient a besoin de plus d'un flacon de TAK-755 par injection,
reconstituer chaque flacon conformément aux instructions mentionnées
dans la rubrique « Reconstitution ». Veuillez noter que le dispositif
BAXJECT II HF est destiné à être utilisé avec un seul flacon de TAK-755
et d'eau pour préparations injectables uniquement, par conséquent, la
reconstitution et l'utilisation d'un deuxième flacon dans la seringue
nécessitent un deuxième dispositif BAXJECT II HF.
- Inspecter visuellement pour détecter la présence de particules et de
décoloration avant l'administration. La solution de TAK-755
reconstituée doit être limpide et incolore.
- Ne pas administrer si des particules ou une décoloration sont observées.
- Administrer TAK-755 dans les 3 heures suivant la reconstitution lorsqu'il est conservé à température ambiante.
- Ne pas administrer TAK-755 dans la même tubulure ou la même poche en même temps que d'autres médicaments injectables.
Reconstitution
1. Préparer une surface plane propre et rassemblez tout le matériel dont vous aurez besoin pour la reconstitution et la perfusion (Figure A).
2. Laisser les flacons de TAK-755 et de solvant revenir à température ambiante avant utilisation.
3. Se laver les mains et les sécher soigneusement.
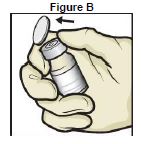
4. Retirer les bouchons en plastique des flacons de TAK-755 et de solvant et placer sur une surface plane (Figure B).
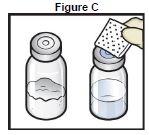
5. Essuyer les bouchons en caoutchouc à l'aide d'un tampon imbibé d'alcool et laisser sécher avant utilisation (figure C).
6. Ouvrir l'emballage du BAXJECT II Hi-Flow en retirant l'opercule, sans toucher l'intérieur (Figure D).
· Ne pas retirer le dispositif BAXJECT II Hi-Flow de l'emballage.
· Ne pas toucher la pointe en plastique transparente

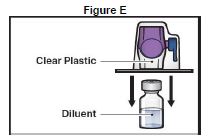
7. Retourner l'emballage avec le dispositif BAXJECT II Hi-Flow et placer au-dessus du flacon de solvant. Enfoncer bien droit jusqu'à ce que la pointe en plastique transparente perce le bouchon du flacon de solvant (Figure E).
8. Saisir l'emballage du dispositif BAXJECT II Hi-Flow par ses rebords et retirer l'emballage du dispositif (Figure F).
· Ne pas retirer le capuchon bleu de l'appareil BAXJECT II Hi-Flow.
· Ne pas toucher la pointe en plastique violet exposée.
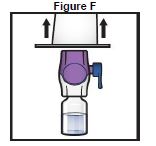
9. Retourner le système de façon que le flacon de solvant soit sur le dessus. Appuyer sur le dispositif BAXJECT II Hi-Flow jusqu'à ce que la pointe en plastique violet perce le bouchon du flacon de poudre deTAK-755 (Figure G). Le vide aspirera le solvant dans le flacon de poudre de TAK-755.
· Vous remarquerez peut-être des bulles ou de la mousse - ceci est normal et devrait disparaître rapidement.
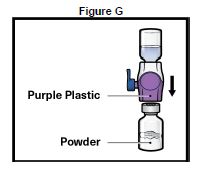
10. Agiter doucement et continuellement les flacons connectés jusqu'à ce que la poudre soit complètement dissoute (Figure H).
· Ne pas secouer le flacon
Figure H
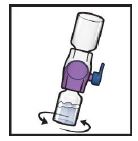
11. Inspecter visuellement la solution reconstituée pour détecter la présence de particules avant administration.
· Ne pas utiliser le produit si des particules ou une décoloration sont observées
12. Si la dose nécessite plus d'un flacon de TAK-755, reconstituer chaque flacon en suivant les étapes ci-dessus.
· Utiliser un autre dispositif BAXJECT II Hi-Flow pour reconstituer chaque flacon de TAK-755 et de solvant.
Instructions d'administration
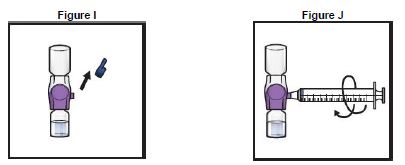
13. Retirer le capuchon bleu du dispositif BAXJECT II Hi-Flow (Figure I). Fixer une seringue Luer- lock (Figure J).
· N'injecter pas d'air dans le système.
14. Retourner le système (le flacon de TAK-755 est maintenant au-dessus). Aspirer la solution reconstituée dans la seringue en tirant lentement sur le piston (Figure K).
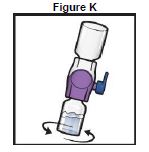
15. Si un patient doit recevoir plus d'un flacon de TAK-755, le contenu de plusieurs flacons peut être aspiré dans la même seringue. Répéter ce processus pour tous les flacons reconstitués de TAK-755 jusqu'à ce que le volume total à administrer soit atteint.
16. Retirer la seringue et fixer une aiguille d'injection appropriée ou un kit de perfusion.
17. Diriger l'aiguille vers le haut et éliminer les bulles d'air en tapotant doucement la seringue avec votre doigt et en poussant lentement et avec précaution l'air hors de la seringue et de l'aiguille.
18. Poser un garrot et nettoyer le site de perfusion avec un tampon imbibé d'alcool (Figure L).
Figure L
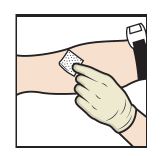
19. Insérer l'aiguille dans la veine et retirer le garrot.
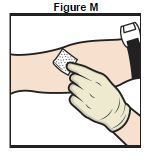
20. Injecter lentement TAK-755 reconstitué, à raison de 2 à 4 mL par minute (Figure M).
· Un pousse-seringue peut être utilisé pour contrôler la vitesse d'administration.
21. Retirer l'aiguille de la veine et exercer une pression sur le site de perfusion pendant plusieurs minutes.
· Ne pas replacer le capuchon sur l'aiguille.
22. Placer l'aiguille, la seringue et les flacons vides dans un collecteur d'objets tranchants
· Ne pas jeter les seringues et les aiguilles dans les ordures ménagères.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation locale en vigueur.
Médicament soumis à prescription hospitalière.
Prescription réservée aux médecins spécialistes en hématologie et aux médecins compétents en maladies du sang.
Médicament nécessitant une surveillance particulière pendant le traitement.
Poudre et solvant pour solution injectable.
Poudre lyophilisée blanche.
Le solvant est une solution limpide et incolore.
TAK-755 1500 UI poudre et solvant pour solution injectable
Chaque boîte contient :
- poudre dans un flacon (verre de type I), muni d'un bouchon en caoutchouc butyle
- 5 mL de solvant dans un flacon (verre de type I), muni d'un bouchon en caoutchouc butyle
- un dispositif de reconstitution (BAXJECT II Hi-Flow)
- une seringue à usage unique de 20 ml
- un kit de perfusion de calibre 25
- deux tampons imbibés d'alcool
TAK-755, 1500 UI poudre et solvant pour solution injectable
Chaque flacon de poudre contient nominalement 1500 UI d'ADAMTS13r.
Après
reconstitution avec 5 mL de solvant (eau pour préparations
injectables), la solution reconstituée a une activité d'environ 300
UI/mL.
TAK-755 est une désintégrine A et métalloprotéinase recombinante à motifs thrombospondine 13 (ADAMTS13r). Il est produit dans des cellules ovariennes de hamster chinois (CHO) par la technologie de l'ADN recombinant.
Pour la liste complète des excipients, voir la rubrique Liste des excipients.
Poudre
Chlorure de sodium
Chlorure de calcium dihydraté
L-Histidine
Mannitol
Saccharose
Polysorbate 80
Solvant
Eau pour préparations injectables